Difference between revisions of "Molten Salt Material"
| Line 3: | Line 3: | ||
=== Results === | === Results === | ||
| − | In this work, we use high-throughput molecular dynamics (HT-MD) to compute thermophysical properties and | + | In this work, we use high-throughput molecular dynamics (HT-MD) to compute thermophysical properties and glean microstructural information about thirty MXn systems of pure molten halide salts exhaustively. These systems include most cations from groups I, II, as well as parts of transition metal elements in lanthanides, and anions from group Ⅶ in the periodic table. Thermophysical properties of these systems, including constant pressure specific heat capacity, density, thermal expansion coefficient, self-diffusion coefficient, viscosity, and microstructure information, which comprises partial radial distribution function and coordination curve under an atmospheric pressure condition, are obtained under a range of temperatures by simulation. These calculations are automated by our own codes called Molten Salt Simulation Toolkit (MSST), developed in the National Supercomputer Center, Guangzhou. Building upon Tianhe-2 high-performance computing (HPC) clusters, MSST can automatically handle input/output processing of CP2K molecular dynamics and manage job submission to cluster queues. Fig. 2 shows the workflow used to implement the HT-MD. (https://raw.githubusercontent.com/pangchq/Molten-Salt-Simulation-Toolkit/master/Fig.2.jpg) |
| − | + | ||
=== Conclusions === | === Conclusions === | ||
Latest revision as of 15:26, 17 June 2021
Contents
Background
Molten salts are promising thermal energy storage materials. The structures and thermophysical properties of pure molten halide salts are essential for complementing the basic thermodynamic data and developing new types of high-performance multi-component halide molten salts. Although the thermophysical properties of multi-components molten halide salts can be roughly estimated by additive principle and other empirical methods without any experiments, the corresponding properties of individual components are momentous in exploiting new compounds. Furthermore, microstructures of molten halide salts need to be simulated and measured to elucidate the evolution law of thermophysical properties under different temperatures. Unfortunately, people could not measure all the thermophysical properties by experiment so far. As an alternative, molecular simulations are proposed and used to predict thermophysical properties over the entire operating temperature range.
Results
In this work, we use high-throughput molecular dynamics (HT-MD) to compute thermophysical properties and glean microstructural information about thirty MXn systems of pure molten halide salts exhaustively. These systems include most cations from groups I, II, as well as parts of transition metal elements in lanthanides, and anions from group Ⅶ in the periodic table. Thermophysical properties of these systems, including constant pressure specific heat capacity, density, thermal expansion coefficient, self-diffusion coefficient, viscosity, and microstructure information, which comprises partial radial distribution function and coordination curve under an atmospheric pressure condition, are obtained under a range of temperatures by simulation. These calculations are automated by our own codes called Molten Salt Simulation Toolkit (MSST), developed in the National Supercomputer Center, Guangzhou. Building upon Tianhe-2 high-performance computing (HPC) clusters, MSST can automatically handle input/output processing of CP2K molecular dynamics and manage job submission to cluster queues. Fig. 2 shows the workflow used to implement the HT-MD. (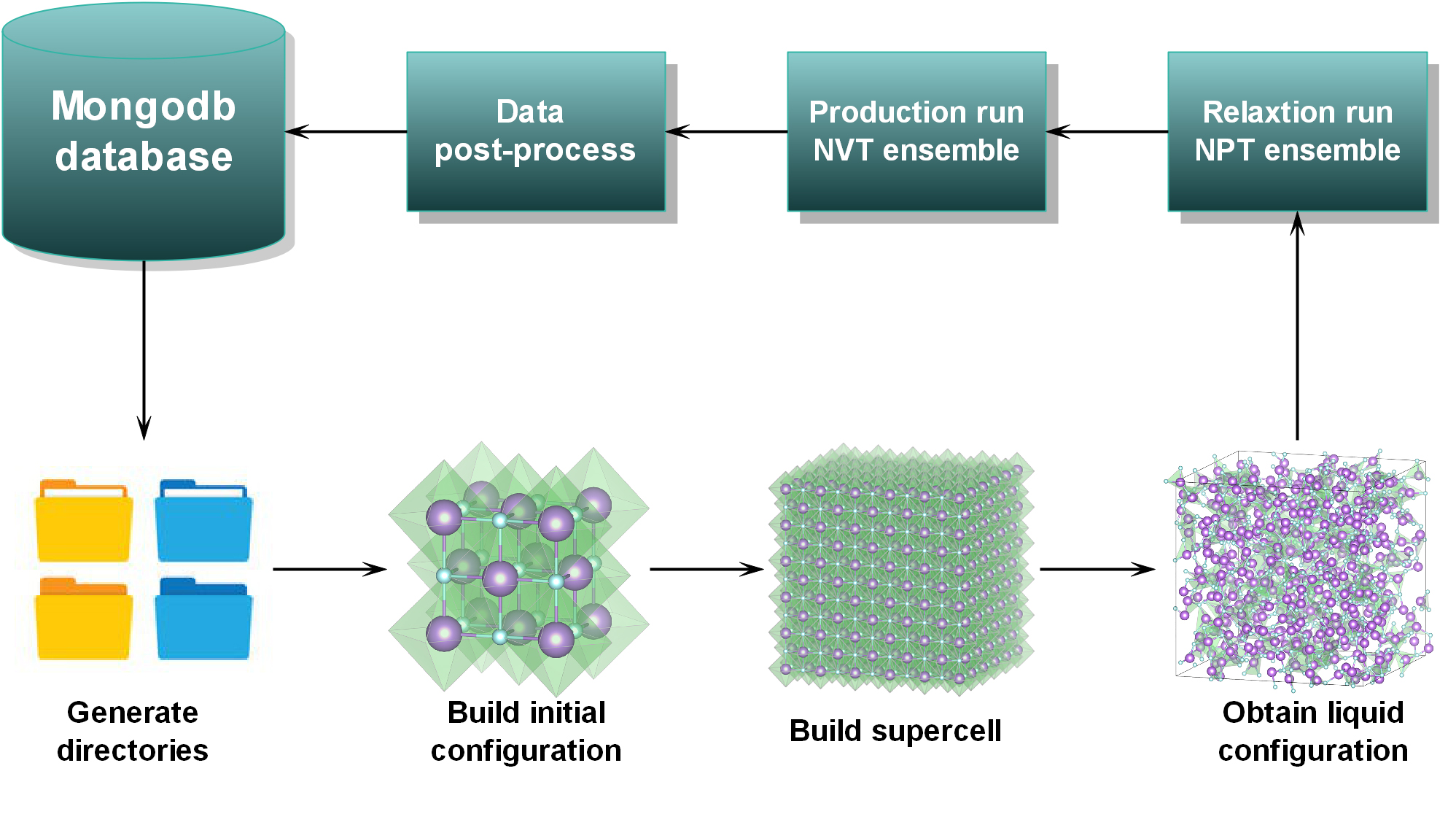 )
)
Conclusions
The simulated constant pressure specific heat capacity, density, viscosity, thermal expansion coefficient, self-diffusion coefficient, and microstructures are in good agreement with experimental values.
Usage notes
We recommend usage of the fitting formula of thermophysical properties in the database as some viscosities of simulations near the melting point are not very accurate due to the reason mentioned above. Researchers who concern the precision and would like to obtain more accurate results can rerun the code (https://github.com/pangchq/Molten-Salt-Simulation-Toolkit/) and increase the simulation time.