Difference between revisions of "Virtual Screening"
(→Job monitoring) |
(→Video Manual) |
||
| (89 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
<div style="margin-left:30px;margin-right:30px"><font size="3" face="TimesNewRoman"><div>__TOC__</div> | <div style="margin-left:30px;margin-right:30px"><font size="3" face="TimesNewRoman"><div>__TOC__</div> | ||
=Virtual Screening Manual= | =Virtual Screening Manual= | ||
| − | VSTH can provide a complete workflow for molecular docking, including PDB file preparation, pocket setting, molecular database preparation, docking program selection, job monitoring, and result analysis and visualization. | + | VSTH can provide a complete workflow for molecular docking, including PDB file preparation, pocket setting, molecular database preparation, docking program selection, job monitoring, and result analysis and visualization.By using this website, you agree that VSTH will not be liable for any losses or damages arising from your use of or reliance on the Content, or other websites or information to which this website may be linked. |
| + | |||
== Target protein preparation == | == Target protein preparation == | ||
| − | <p> | + | |
| − | * | + | <p>During the input file preparation process, the end user can follow the prompts on the web page to provide a structure of the protein target by either uploading a PDB file(1) or providing a PDB code(2). </p> |
| − | * | + | <p>By clicking the "droplist" icon, it will show a detailed list of the selected category. For example, if users clicked the "droplist" icon of protein, all chains of this selected protein will be listed below. By clicking the "delete" icon, users also can delete water, ion, ligand and protein. By clicking the "eye" icon, users can show or hide protein, ligand, water, and ion.</p> |
| − | * | + | What’s more, users can add hydrogen to the target protein by click the "Preprocess" button(3), there are 3 types of adding hydrogen: |
| − | <br><center>http://matgen.nscc-gz.cn/wiki/images/ | + | * add H rotate and flip NQH groups. |
| + | * add H and rotate groups with no NQH flips. | ||
| + | * add H, including His sc NH, then rotate and flip groups. | ||
| + | <br><center>http://matgen.nscc-gz.cn/wiki/images/Vs1.png</center><br> | ||
<p><center><strong>Figure 1.</strong> select protein target screenshot</center></p> | <p><center><strong>Figure 1.</strong> select protein target screenshot</center></p> | ||
<br> | <br> | ||
| − | == | + | == Binding Site Selection == |
| − | <p>VSTH | + | <p>VSTH provides three methods for defining targets and present an interactive viewer of the protein target and the binding site. </p> |
| − | <br><center>http://matgen.nscc-gz.cn/wiki/images/ | + | <p>By default, VSTH will automatically identify pockets using fpocket and select the best scoring pocket as the binding site.The at most 10 ranked pockets are listed and users can get its coordinates and size information. More detailed information is provided when clicking the "Details" button</p> |
| + | <p>Second, users can define the binding site by the centroid of the binding ligand. VSTH will detect ligands in the target protein and show a list of binding ligands for selection.</p> | ||
| + | <p>Third, users can supply the X,Y,Z coordinates of its center and its size directly. </p> | ||
| + | <p>What's more, users can define the number of poses.</p> | ||
| + | <br><center>http://matgen.nscc-gz.cn/wiki/images/Step2.png</center><br> | ||
<p><center><strong>Figure 2. </strong>pocket setting screenshot</center></p> | <p><center><strong>Figure 2. </strong>pocket setting screenshot</center></p> | ||
== Ligand library preparation == | == Ligand library preparation == | ||
| − | <p> | + | |
| + | <p>Available public libraries in VSTH include DrugBank5.0 (approved drug dataset, comprises 2387 molecules), HMDB4.0 (comprises 113,875 molecules) , and InterBioScreen 2020 (purchased nature compound dataset and synthetic compounds dataset vended by InterBioScreen Company,comprises 555,295 molecules). There are many filters available for refining these molecular libraries, including by topological polar surface area, molecular weight, logP, number of acceptors or donors of hydrogen, rotabable bonds and rings. Users can also choose upload their own ligand libraries. VSTH supports compressed files and other file formats like smi, mol2, mol, xyz, cif and sdf, etc. By default, VSTH use the whole DrugBank5.0 as the ligand library.(Fig. 3).</p> | ||
[[Image:vs3.png|800px|frameless|center]] | [[Image:vs3.png|800px|frameless|center]] | ||
| Line 25: | Line 34: | ||
== Docking program selection == | == Docking program selection == | ||
| − | <p>Step 4 allows the user to select docking programs and | + | <p>Step 4 allows the user to select docking programs and set parameters. VSTH provides 6 docking programs (1). Users can select at most 3 programs at one time. Generally, VSTH provides default parameters for docking, while users can modify parameters for personalized docking (2). All advanced parameters are provided according to docking programs and can be found in Table 1. Besides these advanced parameters, users have the option to select DLIGAND2 to re-score the docking conformations. For conformation classification, users can set the cutoff argument (Fig. 4). </p> |
<br><center>http://matgen.nscc-gz.cn/wiki/images/vs4.png</center><br> | <br><center>http://matgen.nscc-gz.cn/wiki/images/vs4.png</center><br> | ||
<p><center><strong>Figure 4. </strong>select docking program and setting parameters</center> </p> | <p><center><strong>Figure 4. </strong>select docking program and setting parameters</center> </p> | ||
<p><center><strong>Table 1. </strong>docking programs and its advanced parameters</center> </p> | <p><center><strong>Table 1. </strong>docking programs and its advanced parameters</center> </p> | ||
| − | <br><center>http://matgen.nscc-gz.cn/wiki/images/ | + | <br><center>http://matgen.nscc-gz.cn/wiki/images/parameters_221017.png</center><br> |
== Job submission == | == Job submission == | ||
| − | <p>On Step 5 | + | <p>On Step 5, users have the option to type in their email. This email address will be used to receive the task id and get reminder when the task has been finished or cancelled.</p> |
| − | [[Image:Vs6.png| | + | [[Image:Vs6.png|800px|center]] |
| − | <p><center><strong>Figure | + | <p><center><strong>Figure 5. </strong>email input screenshot</center></p> |
== Job monitoring == | == Job monitoring == | ||
| − | <p>On | + | <p>On vs_result[https://matgen.nscc-gz.cn/vs_result.html] screen, users have the option to type in their task id, check status, get results and cancel task.To get docking job detail like docking method and binding sites, users can click "Docking Job Details". For those finished jobs, users can click "Download"(1) and get a CSV file which contains a list of filename, score, and cluster id. To download the processed protein file, click the download icon(2). To download the consensus ranking result, click the download icon(3). This consensus ranking list based on the rank of each docking method. For each compounds, let r_i denote the rank of compounds list based on the i_th virtual docking, and ρ_compare denote the product of all ranks. The formula can be expressed as[[Image:consensus_rank.png]]. To download the specific conformation, click the download icon(4) near the conformation 3D viewer.</p> |
| − | [[Image: | + | [[Image:status.png|800px|center]] |
| − | <p><center><strong>Figure 6. </strong> job | + | <p><center><strong>Figure 6. </strong> job status screenshot |
</center></p> | </center></p> | ||
| + | |||
| + | [[Image:Res_page.png|800px|center]] | ||
| + | <p><center><strong>Figure 7. </strong> job result screenshot | ||
| + | </center></p> | ||
| + | |||
| + | =FAQ= | ||
| + | ==Video Manual== | ||
| + | We provide a video manual [https://matgen.nscc-gz.cn/wiki/images/vs2.gif here]. | ||
| + | |||
| + | ==Why can't I get email reminder? == | ||
| + | We recommend users to use academic email. | ||
| + | The enterprise email, like QQ, Yeah, have been tested successfully. | ||
| + | The outlook email is failed to get messages. Other emails require further testing. | ||
| + | |||
| + | ==Why my browser can't display VSTH properly? == | ||
| + | The web interface is written in HTML, JavaScript, Bootstrap and uses 3Dmol.js for displaying. If the browser does not support webgl, some functions may not be available. we recommend users to use browsers like [https://matgen.nscc-gz.cn/wiki/images/vs2.gif Chrome 103.0.5060.66], [https://matgen.nscc-gz.cn/wiki/images/firefox.gif Firefox 101.0.1], [https://matgen.nscc-gz.cn/wiki/images/edge.gif Microsoft Edge 103.0.1264.37], [https://matgen.nscc-gz.cn/wiki/images/opera.gif Opera 91.0.4516.65], [https://matgen.nscc-gz.cn/wiki/images/safari.gif Safari 15.6.1], or above versions, which have been tested. | ||
| + | |||
| + | ==Why can't I get recommended pocket calculated by Fpocket?== | ||
| + | |||
| + | We found that fpocket cannot handle overly large proteins (for instance, The protein of 7LFH contains 80,000 atoms). However, 7LFH is symmetrical with many repeating chains. We recommend that users use the preprocessing tool we developed to delete irrelevant chains. In additional, we have provided more methods for identifying active pocket. Users can identify active pocket by the centroid of the binding ligand, or supply the (X, Y, Z) coordinates of its center and its box size directly. | ||
Latest revision as of 07:06, 18 October 2022
Contents
Virtual Screening Manual
VSTH can provide a complete workflow for molecular docking, including PDB file preparation, pocket setting, molecular database preparation, docking program selection, job monitoring, and result analysis and visualization.By using this website, you agree that VSTH will not be liable for any losses or damages arising from your use of or reliance on the Content, or other websites or information to which this website may be linked.
Target protein preparation
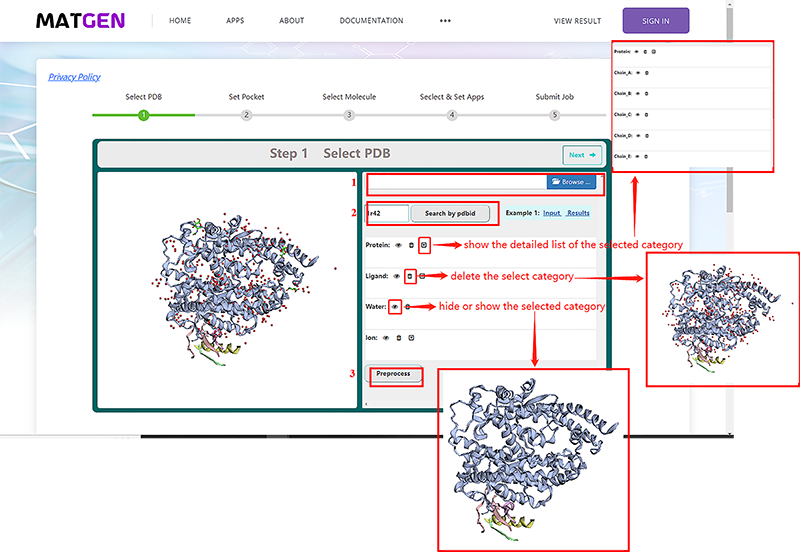
During the input file preparation process, the end user can follow the prompts on the web page to provide a structure of the protein target by either uploading a PDB file(1) or providing a PDB code(2).
By clicking the "droplist" icon, it will show a detailed list of the selected category. For example, if users clicked the "droplist" icon of protein, all chains of this selected protein will be listed below. By clicking the "delete" icon, users also can delete water, ion, ligand and protein. By clicking the "eye" icon, users can show or hide protein, ligand, water, and ion.
What’s more, users can add hydrogen to the target protein by click the "Preprocess" button(3), there are 3 types of adding hydrogen:
- add H rotate and flip NQH groups.
- add H and rotate groups with no NQH flips.
- add H, including His sc NH, then rotate and flip groups.
Binding Site Selection
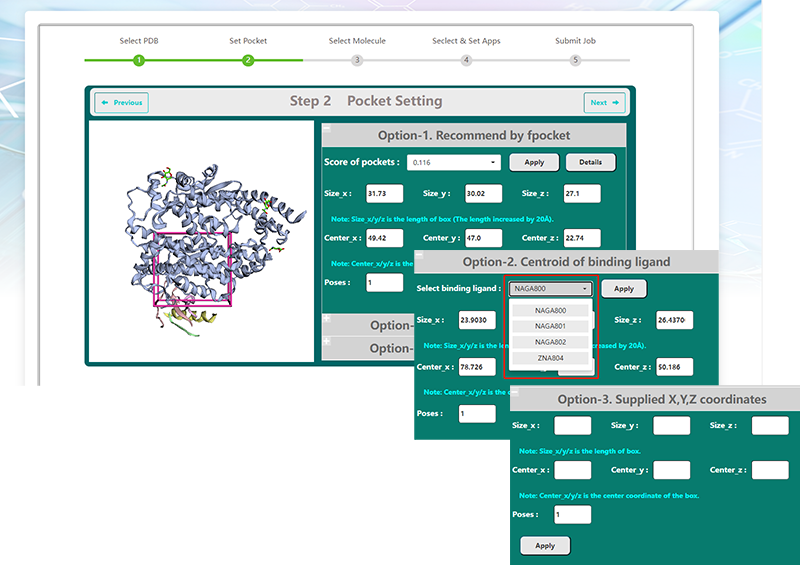
VSTH provides three methods for defining targets and present an interactive viewer of the protein target and the binding site.
By default, VSTH will automatically identify pockets using fpocket and select the best scoring pocket as the binding site.The at most 10 ranked pockets are listed and users can get its coordinates and size information. More detailed information is provided when clicking the "Details" button
Second, users can define the binding site by the centroid of the binding ligand. VSTH will detect ligands in the target protein and show a list of binding ligands for selection.
Third, users can supply the X,Y,Z coordinates of its center and its size directly.
What's more, users can define the number of poses.
Ligand library preparation
Available public libraries in VSTH include DrugBank5.0 (approved drug dataset, comprises 2387 molecules), HMDB4.0 (comprises 113,875 molecules) , and InterBioScreen 2020 (purchased nature compound dataset and synthetic compounds dataset vended by InterBioScreen Company,comprises 555,295 molecules). There are many filters available for refining these molecular libraries, including by topological polar surface area, molecular weight, logP, number of acceptors or donors of hydrogen, rotabable bonds and rings. Users can also choose upload their own ligand libraries. VSTH supports compressed files and other file formats like smi, mol2, mol, xyz, cif and sdf, etc. By default, VSTH use the whole DrugBank5.0 as the ligand library.(Fig. 3).

Docking program selection
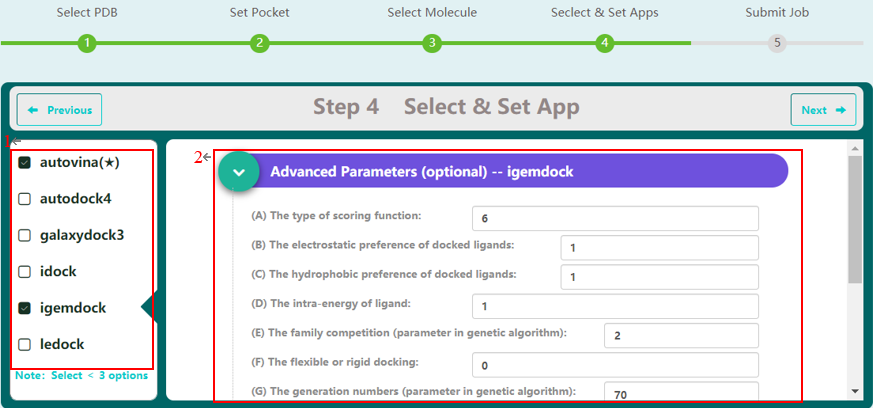
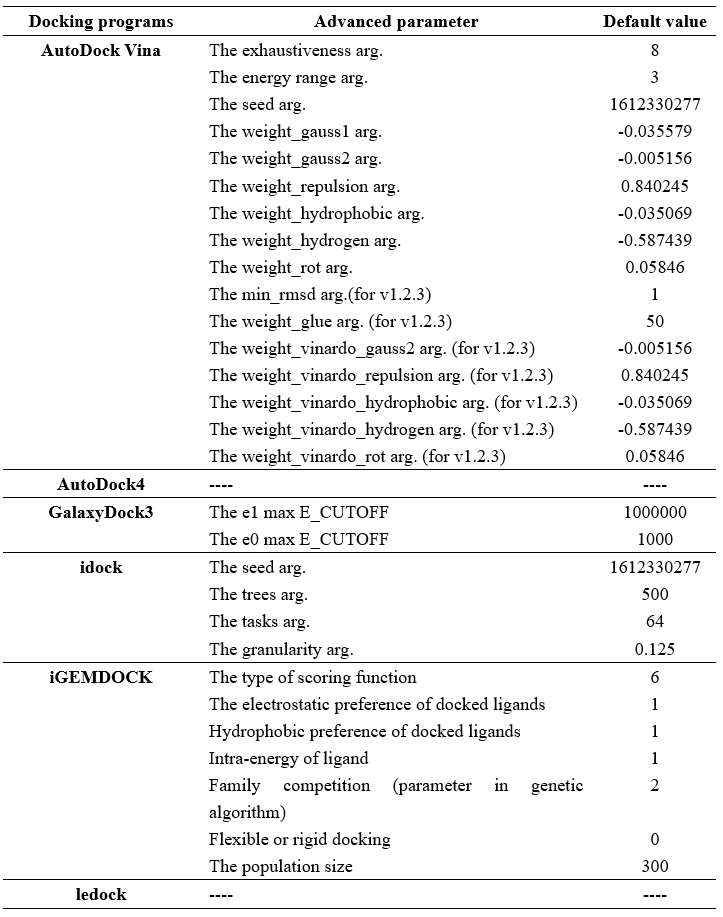
Step 4 allows the user to select docking programs and set parameters. VSTH provides 6 docking programs (1). Users can select at most 3 programs at one time. Generally, VSTH provides default parameters for docking, while users can modify parameters for personalized docking (2). All advanced parameters are provided according to docking programs and can be found in Table 1. Besides these advanced parameters, users have the option to select DLIGAND2 to re-score the docking conformations. For conformation classification, users can set the cutoff argument (Fig. 4).
Job submission
On Step 5, users have the option to type in their email. This email address will be used to receive the task id and get reminder when the task has been finished or cancelled.

Job monitoring
On vs_result[1] screen, users have the option to type in their task id, check status, get results and cancel task.To get docking job detail like docking method and binding sites, users can click "Docking Job Details". For those finished jobs, users can click "Download"(1) and get a CSV file which contains a list of filename, score, and cluster id. To download the processed protein file, click the download icon(2). To download the consensus ranking result, click the download icon(3). This consensus ranking list based on the rank of each docking method. For each compounds, let r_i denote the rank of compounds list based on the i_th virtual docking, and ρ_compare denote the product of all ranks. The formula can be expressed as![]() . To download the specific conformation, click the download icon(4) near the conformation 3D viewer.
. To download the specific conformation, click the download icon(4) near the conformation 3D viewer.


FAQ
Video Manual
We provide a video manual here.
{kind=link}
Why can't I get email reminder?
We recommend users to use academic email. The enterprise email, like QQ, Yeah, have been tested successfully. The outlook email is failed to get messages. Other emails require further testing.
Why my browser can't display VSTH properly?
The web interface is written in HTML, JavaScript, Bootstrap and uses 3Dmol.js for displaying. If the browser does not support webgl, some functions may not be available. we recommend users to use browsers like Chrome 103.0.5060.66, Firefox 101.0.1, Microsoft Edge 103.0.1264.37, Opera 91.0.4516.65, Safari 15.6.1, or above versions, which have been tested.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Why can't I get recommended pocket calculated by Fpocket?
We found that fpocket cannot handle overly large proteins (for instance, The protein of 7LFH contains 80,000 atoms). However, 7LFH is symmetrical with many repeating chains. We recommend that users use the preprocessing tool we developed to delete irrelevant chains. In additional, we have provided more methods for identifying active pocket. Users can identify active pocket by the centroid of the binding ligand, or supply the (X, Y, Z) coordinates of its center and its box size directly.