Virtual Screening
Virtual Screening Manual
We provide a complete workflow for molecular docking, including target protein preparation, pocket setting, molecular database preparation, docking program selection, job submission, job monitoring, and result analysis and visualization.
target protein preparation
During the target protein preparation process, the end user can follow the prompts on the web page to provide a structure of the protein target by either providing a 4-character PDB accession code (1) or by uploading a PDB file (2). Users also can delete water, ion, ligand and protein (3) as they want. What’s more, users can add hydrogen to the target protein (4), there are 3 type of adding hydrogen, add H and rotate and flip NQH groups; add H and rotate groups with no NQH flips and add H, including His sc NH, then rotate and flip groups.

pocket setting
MDWS will automatically identify pockets using fpocket [9] and present an interactive viewer of the protein target and the best scoring pocket (1). Detail information on the provided target (2), such as druggability score, number of alpha spheres, total SASA and so on, can also be found on this screen. Users can select the top 10 identified pockets by fpocket or change the box size and center for docking (3). What’s more, users can define the number of poses (4) which docking programs will use to generate conformations on this screen.

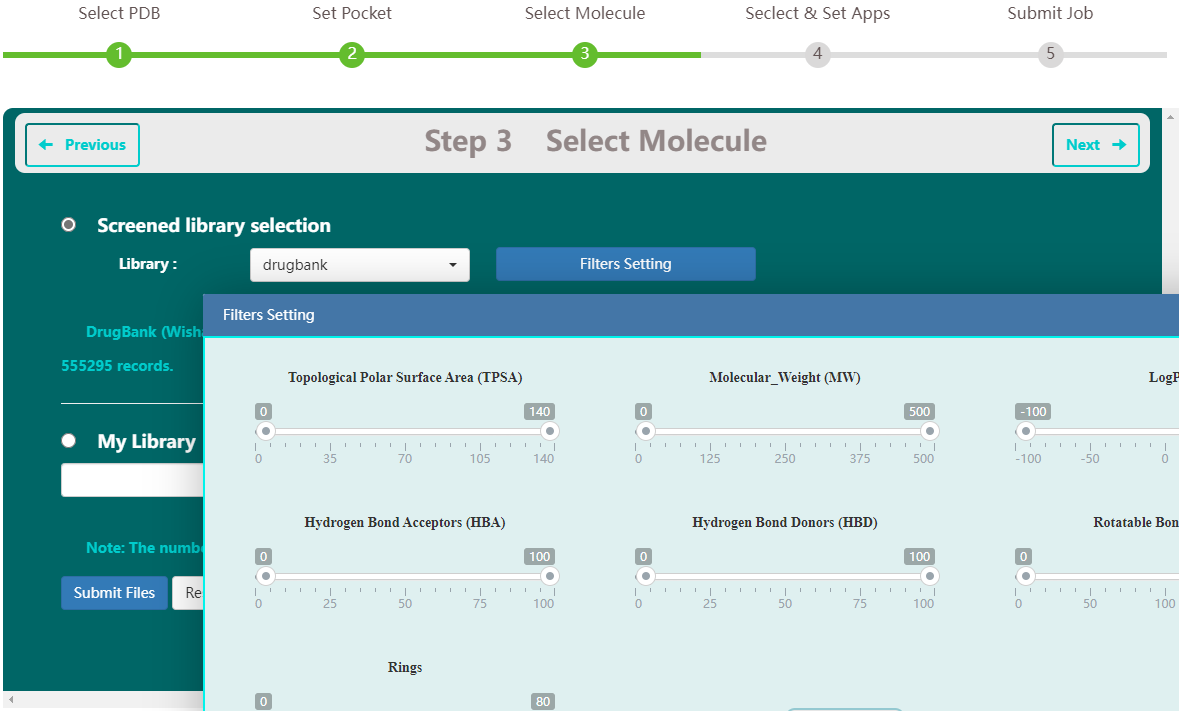
molecular database preparation
Step 3 allows the user to select libraries for virtual screening on MDWS. MDWS provides one public ligand library and one test ligand library. The data of the public library is selected randomly from ZINC15 (Stering, et al, 2015) and 1000 records are included. Users can also upload their own ligand library. MDWS supports compressed files, such as zip file, tar file, and so on. Also, MDWS uses OpenBabel (O’Boyle et al., 2011) for format transformation, so VSTH supports formats like smi, mol2, mol, xyz, cif and sdf.
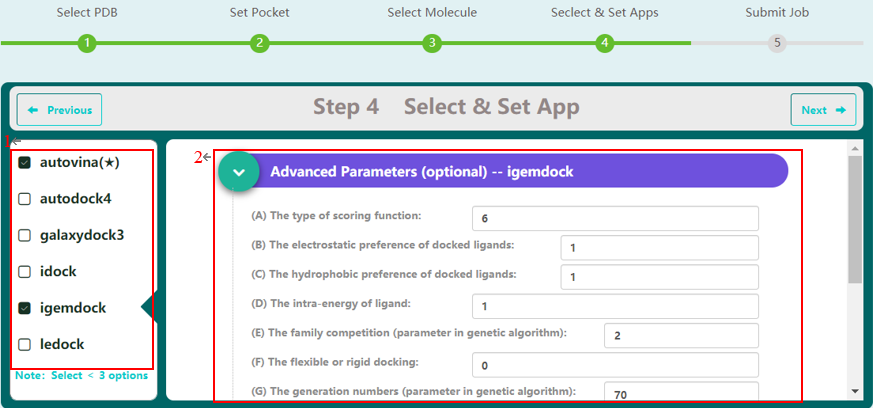
docking program selection
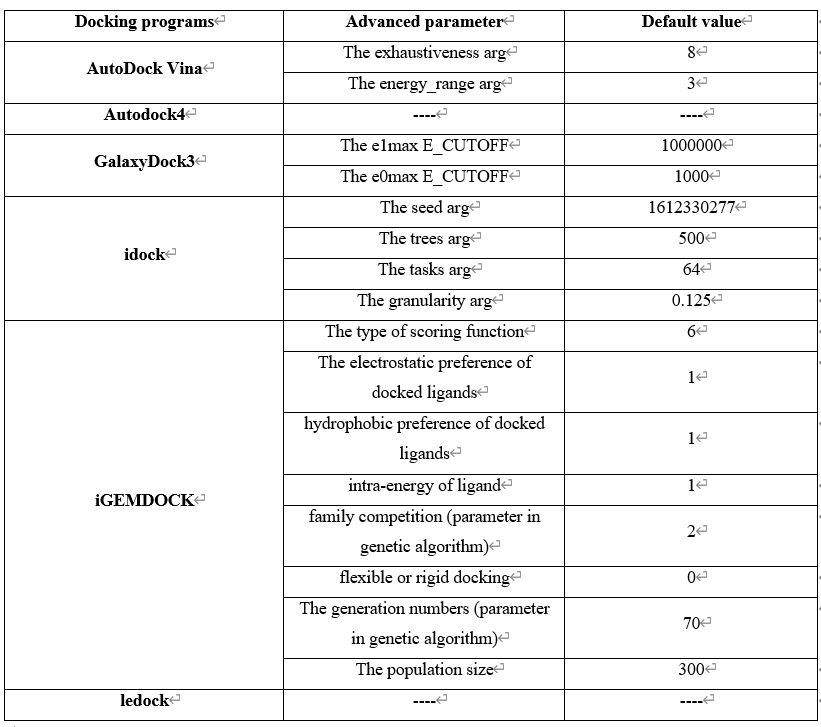
Step 4 allows the user to select docking programs and setting parameters for virtual screening on MDWS. MDWS provides 6 software for docking (1). Users can select more than 1 program for docking. Generally, MDWS provides default parameters for docking, but users can modify parameters related to docking (2), such as the type of scoring function, the electrostatic preference of docked ligands, on this screen. All advanced parameters are provided according to docking programs and can be found in Table 2. Besides these advanced parameters, users have the option to select DLIGAND2 to predict protein-ligand binding affinity.
job submission
On Step 5 screen, users have the option to type in their email. This email address will be used to receive taskid. This taskid is a unique identifier, and users need it in the job monitoring process to check its status and get results.
job monitoring
<p>On Step 6 screen, users have the option to type in their jobid, check status and get results.</p>
<center> </center>
</center>
<p><center>Figure 7. job monitoring screenshot
</center></p>